

The European Health Technology Assessment (HTA) landscape presents many potential challenges for pharmaceutical manufacturers seeking reimbursement of new, innovative products.
However, these barriers to market access can be anticipated by examining and understanding the stringent evidence requirements that are in place within each system. Some of the factors that negatively impact a new product's evaluation include open-label or non-comparative trial designs, including the use of trial comparators that are not standard of care, or endpoints that may not be considered patient relevant. By understanding these requirements and taking them into account when designing clinical trials, manufacturers can improve their chances of securing market access for innovative products in European markets.
Why are manufacturers not meeting European payer evidence requirements in HTA evaluations?
There are several reasons why a manufacturer may not appropriately account for a country's evidence requirements prior to HTA. Firstly, manufacturers may deem the process of generating the appropriate evidence to be too costly or may see head-to-head trials as too risky in case superiority cannot be demonstrated. Clinical trials are often designed with regulatory requirements in mind or to meet the needs of US payers, meaning European payer requirements are often a secondary consideration. This can be a result of prioritizing the US market because of the potential for premium prices and lucrative commercial prospects, late involvement of non-US market access teams and/or a lack of forward thinking about the wider pricing and access implications.
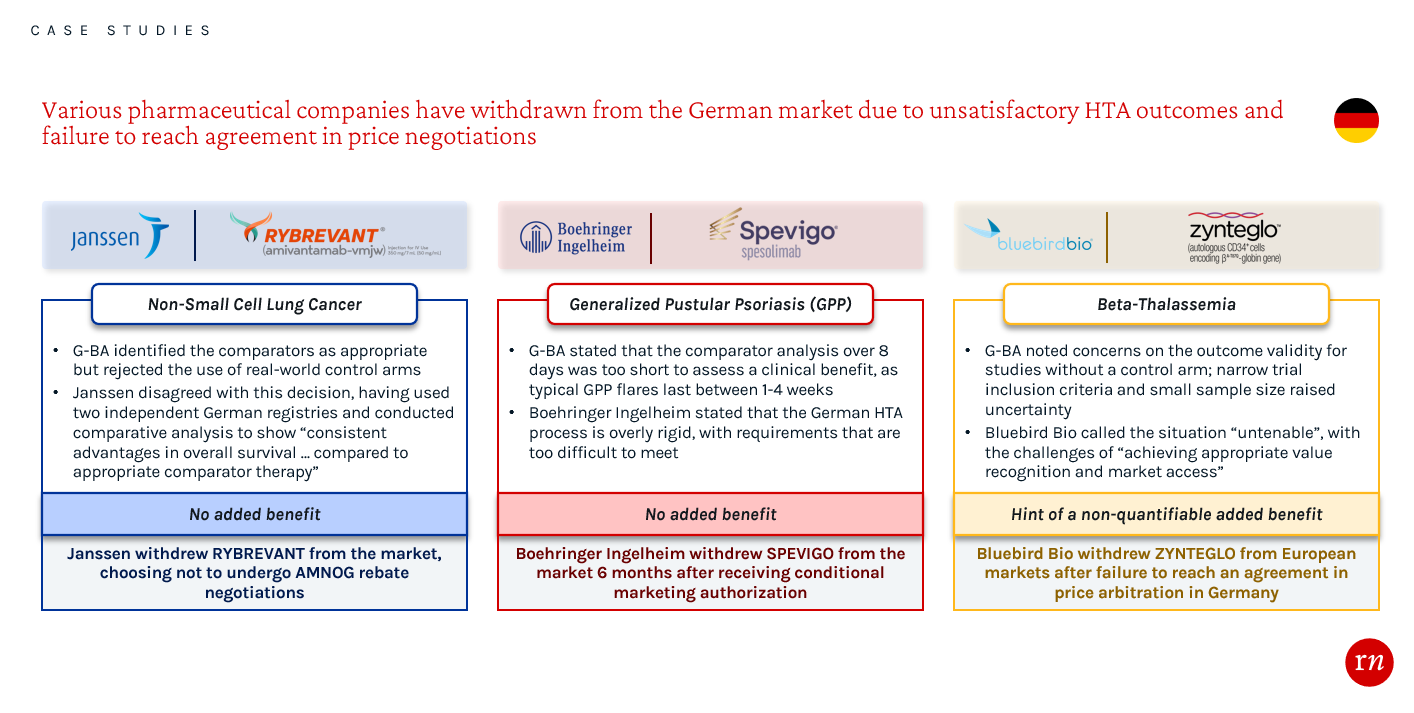
Overlooking EU evidence requirements generally leads to a less favorable HTA evaluation and hinders manufacturers’ leverage during approval and price negotiation processes. Not meeting the strict evidence requirements in Germany has led to a number of recent product withdrawals from the market. These examples highlight the importance of conducting thorough assessments while planning and executing clinical trials, as well as during the process of preparing evidence packages. By engaging with payers early about the evidence requirements in Germany, companies may have been able to receive a favorable HTA and pricing outcome.

What changes are manufacturers making to their product planning, trial design or evidence generation strategies and organizations?
Some pharmaceutical manufacturers are starting to devise strategies to mitigate the implications of not adhering to the evidence requirements of European payers. Firstly, companies are embracing earlier ‘go/no go’ decisions, allowing for higher investment and allocation of resources to the most promising products to ensure they have sufficient backing and that these evidence requirements are met. Furthermore, companies are redirecting their focus towards markets with comparatively less stringent evidence requirements such as China. With times to reimbursement reducing and receptiveness to pharmaceutical innovation increasing, China is an increasingly attractive market for manufacturers.
Within pharmaceutical organizations, understanding of the importance of market access is growing, as seen through the creation of ‘Chief Access Officers’ in some organizations, with the view of ensuring that market access is considered through all stages of drug development. Work is also being done to ensure the role of market access is understood across all functions through ‘market access academies’ — internal training programs to educate broader teams on the importance of market access across the product lifecycle and ensure market access is top of mind so that requirements of payers from all markets are met.
What are the potential solutions when a manufacturer is unable to meet the evidence requirements?
Modeling
In some markets such as the UK, Spain and Italy, statistical modeling is an accepted approach to mitigate lack of head-to-head data or relevant trial outcomes. Indirect treatment comparisons (ITCs) can be used to compare treatments to standard of care. However, for ITCs to be accepted in Germany, they must meet the high methodological requirements set by the G-BA. Surrogacy analyses are also an option to help demonstrate the patient relevance of biological markers but, again, will only be accepted by certain markets and must be robust. For example, through modeling and clinician opinion, NICE accepted that short-term weight loss and improved diabetic status were acceptable surrogates for reduced long-term cardiovascular risk during the review of liraglutide for managing obesity.
Restricting the patient population
One alternative method of overcoming the challenges presented by European markets’ rigorous evidence requirements involves strategically restricting the reimbursed population, or even the indication, to patients for whom the drug is particularly effective. Whilst this strategy can help a manufacturer ensure that its product achieves the highest possible pricing potential, it means sacrificing patient volume. It is therefore vital for manufacturers to understand the price-volume trade off and its impact on global revenue.
Leverage the patient voice
Manufacturers can also intensify their lobbying efforts and leverage the perspectives of patients to ensure favorable evaluation. By presenting testimonials or narratives from affected patients, as well as clinicians, manufacturers can highlight the unmet need that would be addressed by their product and apply pressure on European payers to grant approval for their product, even if it may not fully meet evidence standards.This was the case in November 2022, when olaparib was provisionally rejected for use on the NHS in England. However, through the Olaparib Now campaign initiated by the patient organization, Breast Cancer Now, 70,000 signatures were collected urging the manufacturer, NICE and NHS England to come to an agreement to make olaparib available to patients. This led to the reversal of NICE’s provisional decision and recommendation for use of Olaparib within the Cancer Drugs Fund.
Real-world evidence
If a traditional or comparative clinical trial is not feasible for a manufacturer, there are some opportunities to collect real-world evidence to corroborate a product’s efficacy and safety data. For some ultra-rare or life-threatening conditions with no existing treatments, where placebo-controlled trials are not feasible, retrospective natural history data can be used as a comparator and are generally accepted by payers. For example, selumetinib is reimbursed in France, Germany, Spain and the UK for neurofibromatosis type 1 based on a Phase 2 trial with an external control arm comprising natural history data and a previous clinical trial for a different drug. There are also opportunities to collect additional data from early access or compassionate use programs, which can supplement clinical data and will be considered by payers in the UK and France. However, manufacturers should prepare for the possibility that not all HTA bodies, such as the G-BA in Germany, will accept real-world evidence as an appropriate demonstration of a product’s efficacy and safety. Speaking with payers and reviewing past applications can help manufacturers to understand how HTA bodies may judge the real-world evidence they plan to present.
Conditional approval
Where there is uncertainty on the clinical benefit of a drug, manufacturers may also utilize real-world evidence to achieve optimal access and reimbursement through conditional approval. Additional efficacy and safety data are collected post launch and, after an agreed length of time, used to support re-evaluations of reimbursement and price. Conditional approval is an option in the UK through the Cancer Drugs Fund or Innovative Medicines Fund. More recently, Germany have introduced the requirement for select orphan drugs, where there is uncertainty in the data, to collect post-launch real-world evidence for an agreed time period, which is followed by a price renegotiation. This was recently agreed for BioMarin’s Roctavian (valoctocogene roxaparvovec-rvox) for hemophilia A, for which additional data on mortality, morbidity, health-related quality of life and side effects will be collected over 3 years. Conditional approval mechanisms ensure continuous assessment and fair pricing of pharmaceuticals, benefiting both payers, manufacturers and patients.
What is the impact of the EU JCA?
The EU JCA is due to take effect in January 2025 and is intended to create greater efficiencies for both HTA bodies and pharmaceutical companies, as well as greater access for patients in lower-income European countries. Published methods suggest that evidence requirements will remain high with a strong preference for head-to-head data from randomized controlled trials. However, due to differing economic requirements and review processes, national HTA evaluations will not be completely replaced meaning that individual markets can continue to set their own criteria on what evidence they need to see.
Conclusion
There are many reasons why manufacturers do not meet the stringent evidence requirements set by EU payers, but there are also numerous solutions available to companies to ensure timely access to their products at an acceptable price. Early consideration of EU HTA requirements when creating evidence generation plans is key to launch success. The sooner market access experts are involved, the better placed manufacturers are to foresee and mitigate unexpected challenges during the reimbursement process. Obtaining early payer feedback on evidence generation plans, either through early scientific advice from HTA bodies, advisory boards or interviews, can help identify areas of concern so mitigation strategies can be put in place. Ultimately, proactive payer engagement and early collaboration with market access stakeholders can pave the way for smoother product launches and improved access for patients in need of innovative treatments.
By Isobel Owens-Smith, Consultant, Red Nucleus Market Access & Commercialization.
Contact us to find out more about how our Market Access and Commercialization Services team can support you with your evidence generation strategy.